La enfermedad de Huntington, una enfermedad degenerativa y silenciosa
Por Adriana Arango-Velez Ph.D.
Una enfermedad que no es tan conocida como el Parkinson o Alzheimer, pero que de igual manera afecta las células del cerebro causando atrofia. La patología de la enfermedad de Huntington es causada por una única mutación genética que resulta en una versión anormal de la proteína “Huntingtina” (HTT), induciendo así la sobre-producción y acumulación de esta proteína en el cerebro [1]. Aunque nuestras células son capaces de descomponer y reciclar el exceso de proteínas, mediante la autofagia (proceso natural de reciclaje que ocurre en las el lisosoma de las células [2]); al ocurrir la mutación del gen HTT, se produce una proteína defectuosa, que no puede ser completamente degradada por la autofagia, formando fragmentos de proteínas toxicas que se pegan y acumulan dentro de las células. Debido a que nuestras neuronas son sensibles a la agregación de proteínas, una sobre acumulación de éstas es perjudicial para el funcionamiento neuronal, llevando a la degeneración de células nerviosas, y en ultima instancia a la muerte celular [3; 4]. Los síntomas iniciales son cambios en la personalidad haciendo a la persona agresiva, violenta, ansiosa y deprimida. Ya que esta enfermedad ataca las células nerviosas afecta también el movimiento físico y la capacidad de discernir. Es una enfermedad hereditaria con un 50% de posibilidad de ser adquirida, y silenciosa ya que aparece entre los 30 y 50 años de edad [1]. En Colombia se han detectado varios casos en el Magdalena, Juan de Acosta en el Atlántico, Antioquia, Choco, Medellín y Bogotá [5; 6]; sin embargo, no se sabe cuanta gente tiene esta enfermedad en Colombia [6].
El descubrimiento y causa de la enfermedad de Huntington
Esta enfermedad fue descrita inicialmente en 1872 por George Huntington. Debido a que esta enfermedad se manifiesta después de los 30 años, la mayoría de las personas afectadas ya han tenido hijos y han pasado la mutación del gen HTT a la siguiente generación. En familias numerosas, la enfermedad puede aparecer en cada generación debido a su condición autosómica dominante, lo que significa que en organismos diploides como nosotros en los que existen dos copias de cada uno de nuestros genes, basta heredar de alguno de los progenitores una copia alterada para desarrollar la enfermedad. En la Fig. 1, se representa el caso de una familia americana en el cual la enfermedad se presenta en cinco generaciones [1;7]. Aunque los síntomas de la enfermedad casi siempre aparecen en la edad adulta, la mutación dominante del gen HTT esta presente desde el nacimiento [1].
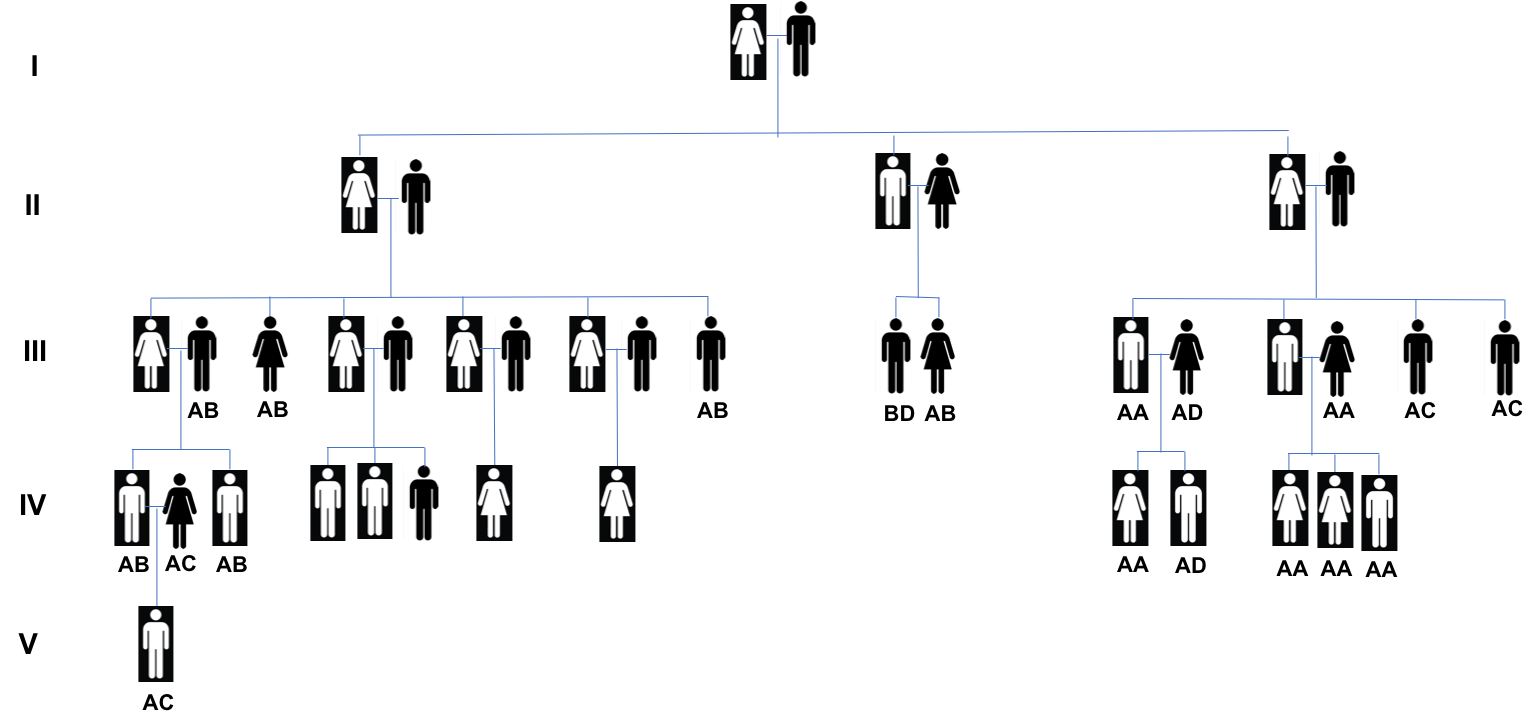
Figura 1. Cinco generaciones de una familia afectada por la enfermedad de Huntington. Cuadros en negro son las personas que poseen el gen dominante HTT mutado que produce la enfermedad. Adoptado de Gusella et al. 1983 y Chial, H. 2008 [1,7].
El gen normal de huntingtina contiene entre 10 a 35 repeticiones de la secuencia citosina, adenina y guanina (CAG) en la región codificante del gen. Cuando esta repetición CAG se repite más de 40 veces ocurre la alteración disfuncional del gen, debido al alargamiento de poliglutaminas (o polyQ – porción de una proteína conformada por varias unidades de glutamina-) [5;8]. Al alargarse la región de CAG en el exón del RNA mensajero del HTT, ocurre toxicidad debido a que las perturbaciones que se presentan en la expresión génica [9].
Históricamente esta enfermedad ha sido mas frecuente en personas de raza blanca, ya que se especula que la enfermedad se inicio en el Norte Europeo y fue llevada posteriormente a diferentes partes del mundo por los inmigrantes colonos europeos [8]. Sin embargo, se han reportado varios casos en Latinoamérica (México, Venezuela, Perú, Chile, Brasil y Colombia) [5; 10], algunos países del continente africano, China, India [11], e Italia [12].
Los avances científicos en el genoma humano han permitido identificar por medio de exámenes genéticos, personas que poseen el gen mutado mucho antes de que los síntomas se presenten. Además de un examen genético de ADN para descubrir si una persona posee esta enfermedad, hay otros métodos utilizando imágenes neurológicas que pueden mostrar fluidos en el cerebro, degeneración cortical, atrofia cerebral, o exceso de materia gris o perdida de materia blanca. Para mas información en este aspecto ver Paulsen, 2010 [13]. Aunque no existe cura para esta enfermedad, existen estudios clínicos para tratar de silenciar el gen que codifica para la proteína mutada utilizando la técnica de siRNA, además de la utilización de células madre mesenquimales [14] y oligonucleótidos antisentido. Brevemente, las técnicas de siRNA (de sus siglas en ingles small interfering RNA), y de oligonucleótidos antisentido, se basan en la unión de un oligonucleótido a un RNA objetivo para neutralizarlo, degradándolo o impidiendo su traducción a proteína [15]. Las células madre mesenquimales son células multipotenciales primitivas que se originan a partir de la capa germinal y tienen la capacidad de diferenciarse en diversos tipos de células [16]. Estas células se utilizan en la terapia celular para reemplazar las células disfuncionales o células muertas producidas por la enfermedad de Huntington [14]. Adicionalmente, existen varias investigaciones usando la técnica de CRISPR/Cas9 (una tecnología de edición génica) para silenciar el gen HTT mutado y prevenir la formación de la proteína defectuosa. En este proceso, se utiliza la enzima Cas9, la cual corta el ADN de manera precisa mediante el uso de una guía de ARN complementario a la secuencia genética de la región mutada del gen HTT, silenciando su expresión [17; 18]. Estas terapias génicas están aun en proceso de estudio utilizando células humanas y animales para evaluar su eficacia.
A pesar de existir nuevas tecnologías para detectar y controlar la enfermedad de Huntington, los casos reportados en Colombia son de familias quienes no cuentan con los recursos económicos suficientes para someterse a un tratamiento ya sea genético, o pagar el precio de drogas que puedan disminuir los síntomas de la enfermedad. Para mayor información acerca del estado y los avances de esta enfermedad en Colombia visitar Fundación Huntington Colombia [19].
Referencias
[1] Chial, H. 2008. Huntington’s disease: The discovery of the Huntingtin gene. Nature Education 1(1);71
[2] Wikipedia. Autofagia. https://es.wikipedia.org/wiki/Autofagia
[3] Pircs, K., Petri R., Madsen S., et al. 2018. Huntingtin Aggregation Impairs Autophagy, Leading to Argonaute-2 Accumulation and Global MicroRNA Dysregulation. Cell Reports 6, 1397-1406. https://www.cell.com/cell-reports/fulltext/S2211-1247(18)31107-0?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS2211124718311070%3Fshowall%3Dtrue
[4] Cortes C.J. La Spada, A.R. 2014. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov.Today 19, 963-971.
[5] Huntington Disease in South America (Blog) http://web.stanford.edu/group/hopes/cgi-bin/hopes_test/south-america/#resources
[6] Huntington’s disease: the new gene therapy that sufferers cannot afford. https://www.theguardian.com/science/2016/may/15/huntingtons-disease-drugs-cure-research-poor-families-colombia-corporate-responsibility
[7] Gusella J. F.; Wexler, N.S.; Coneally, P.M.; Naulor, S.L. et al. 1983. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 235.
[8] Kremer B.; Goldberg P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Bassett, A.; Almqvist E. et al. 1994. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med, 330(20);1401-6.
[9] Marti E. 2016. RNA toxicity induced by expanded CAG repeats in Huntington’s disease. Brain Pathol 26, 779-786.
[10] Arango-Lasprilla, J.C.; Iglesias-Dorado, J.; Moreno S; Lopera F. 2003. A neuropsychological study of Huntington’s disease in families in Antioquia, Colombia. Rev Neurol, 37(1):7-13.
[11] How many people have Huntington disease? https://huntingtonstudygroup.org/hd-insights/how-many-people-have-huntington-disease/
[12] Carrasi E., Pugliatti, M., Govoni V., Sensi M., Casetta I., Granieri E. 2017. Epidemiological Study of Huntington’s disease in the province of Ferrara, Italy. Neuro epidemiology, 49:18-23.
[13] Paulsen, J.S. 2010. Early detection of Huntington disease. Future Neurol 5(1):10.2217/fni.09.78 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3860286/
[14] Wooseok I., Soon-Tae L., Kon C., Manho K., Jae-Kyu R (2009). Stem cells transplantation and Huntington’s disease. Int J. Stem Cells 2(2): 102-108.
[15] Watts, J.K.; Corey, D.R. 2014. Gene silencing by siRNAs and antisense oligonucleotides in the laboratory and the clinic. J. Pathol. 226(2):365-379 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3916955/
[16] Wikipedia. Celula madre mesenquimatosa https://es.wikipedia.org/wiki/C%C3%A9lula_madre_mesenquimatosa
[17] Dabrowska, M. Juzwa, W., Krzyzosiak W.J., Olejniczak M. 2018. Precise excision of the CAG tract from the Huntingtin gene by Cas9 Nickases. Front. Neurosci. https://doi.org/10.3389/fnins.2018.00075.
[18] CRISPR Reverses Huntington’s disease in mice. Genetic Engineering & Biotechnology News. https://www.genengnews.com/topics/translational-medicine/crispr-reverses-huntingtons-disease-in-mice/
[19] Fundación Huntington de Colombia. http://fuhcol.blogspot.com/2014/12/investigaciones-recientes-sobre-la.html#comment-form
Comentarios